
Az Európai Bizottság azt reméli, hogy a klinikai vizsgálatokról szóló 536/2014/EU Rendelet (Clinical Trials Regulation=CTR) bevezetésével vonzó és kedvező környezetet biztosít az EU-n belüli klinikai vizsgálatok lefolytatásához. [2], [3] 2023. január 31-ével kezdődően a CTR az az új uniós szabályozás, amelyet be kell tartani minden, az EU-ban emberi felhasználásra szánt gyógyszerekkel végzett intervenciós klinikai vizsgálati kérelem során. [1], [2] Célja a benyújtási és értékelési folyamatok harmonizálása, a tagállamokon belüli és a tagállamok közötti együttműködés javítása magas szintű nyilvános átláthatósággal, magas szintű betegbiztonsági előírások mellett. [2]
Ebben az összefoglalóban áttekintjük a klinikai vizsgálatokról szóló 2001/20/EK Irányelv (Irányelv) és a CTR (Rendelet) közötti közötti kulcsfontosságú változásokat és azt, hogy az átmeneti időszakban milyen kihívásokkal szembesülhetünk. [3], [4]
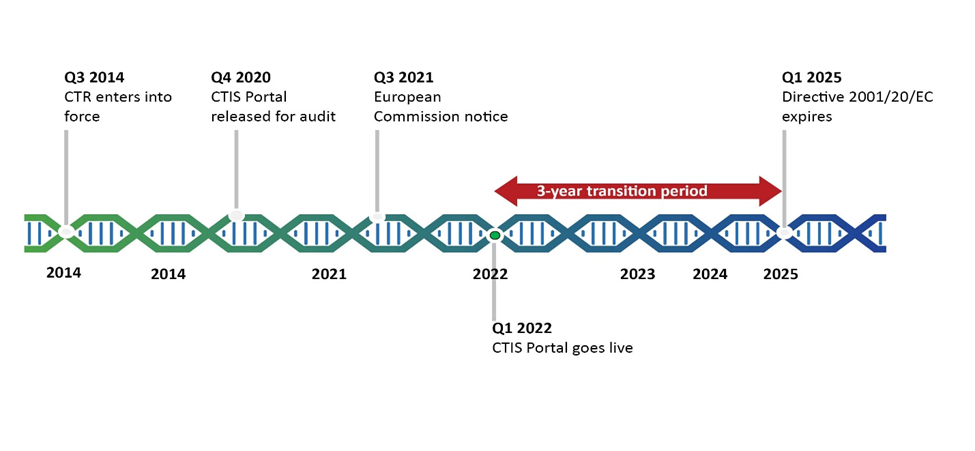
2004 előtt a klinikai vizsgálatok folyamatait és követelményeit tagállami szinten határozták meg, ezért az EU-országok között nagy eltérések mutatkoztak. 2004-ben elfogadták az Irányelvet, amely az első lépés volt a klinikai vizsgálati folyamatok és követelmények harmonizációja felé. Az Irányelv értelmében azonban a klinikai vizsgálatok alkalmazása továbbra is rendkívül nagy különbözőséget mutatott, az egynél több tagállamot érintő klinikai vizsgálatokat minden egyes országban külön kérelmezték. A CTR-ral még egy lépéssel közelebb kerültünk a tagállamok közötti harmonizációhoz. [2]
A CTR bevezette a CTIS (=Clinical Trials Information System) portált, amely lehetővé teszi az összes klinikai vizsgálati kérelem egyetlen rendszeren történő benyújtását. Ezen a rendszeren keresztül a több tagállamot érintő klinikai vizsgálatok kérelmét a megbízók egyetlen kérelemként nyújtják be az összes országra vonatkozóan. A kérelmet a tagállamok közösen értékelik és egyetlen határozatot hoznak a vizsgálat Part I részét tekintve. A Part I rész a vizsgálat általános, szponzori és termékinformációit tartalmazza. [2]
A Part II rész, amely egy-egy tagállam nemzeti elvárásai szerinti dokumentációit tartalmazza és tagállami szinten kerül értékelésre, szintén a CTIS rendszeren keresztül kerül benyújtásra. Egy-egy klinikai vizsgálati kérelem engedélyezéséhez mind a Part I, mind a Part II dokumentációjának jóváhagyása szükséges. Az Európai Gyógyszerügynökség (European Medicines Agency=EMA) közzéteszi a portálra feltöltött dokumentumokat, ezzel is növelve a klinikai vizsgálatokra vonatkozó információk átláthatóságát. [2]
2022. január 31-én a CTIS rendszer életbe lépett. Ettől a naptól kezdve lehetőség nyílt az új klinikai vizsgálati kérelmek portálon keresztüli benyújtására, viszont kötelezővé 2023. január 31-én vált. A CTR-re történő átállás megkönnyítése érdekében a megbízók számára lehetővé tették, hogy egy 3 éves átmeneti időszakban a még Irányelv szerint engedélyezett klinikai vizsgálatokat továbbra is az Irányelv szerint folytassák le. [1]
Azonban 2025. január 31-től az Irányelv alapján jóváhagyott, továbbra is folyamatban lévő vizsgálatoknak meg kell felelniük a CTR-nek, és megbízóiknak a CTIS-ben fel kell vezetni az ezekre vonatkozó információkat és dokumentumokat. Az EMA arra ösztönzi a megbízókat, hogy használják ki az átmeneti időszakot és a klinikai vizsgálatokra vonatkozó információikat időben rögzítsék a CTIS-ben. [1]
Az MSD Pharma Hungary Kft. (MSD) megbízói oldalról az elsők között regisztrált a CTIS rendszerben. 2022. májusban az első, korábban az Irányelv szerint engedélyezett klinikai vizsgálat rendeletre történő átvezetési („transition trials”) kérelme benyújtásra került a CTIS rendszeren keresztül, majd 2022. szeptemberében az első, a rendelet szerinti új klinikai vizsgálati-kérelem beadása is megtörtént.
„Early adopter”-ként a CTIS életbe lépése után rövid időn belül bátran éltünk a korai tapasztalatszerzési lehetőséggel, ugyanakkor az új rendszer és a szabályozás számos kihívás elé állította vállalatunkat és jó néhány kérdést azonosítottunk. A kihívások rámutatnak a CTR több pontját érintő általános megfogalmazásra (pl. beteganyagok – betegek kezéhez kerülő anyagok, „transition” vizsgálatok kérelméhez benyújtandó dokumentum-elvárások, CTIS használata). A kérdések tisztázását segítendő az EMA Query Management munkacsoportja rendszeresen kiadja a Q&A dokumentum egy-egy újabb verzióját, amelyben válaszol a szponzori szervezetektől öszegyűjtött, a CTIS használatával és a klinikai vizsgálatokról szóló rendelettel kapcsolatos főbb kérdésekre. [3]
Mindemellett a vizsgálatvezetők részére új, kötelezően kitöltendő formanyomtatványok is bevezetésre kerültek („Investigator CV template”, „Site Suitability Template”), amelyeket az ETT KFEB az EMA formanyomtatványok alapján a magyar elvárások szerint alakított ki. A formanyomtatványok folyamatos változásának követése és alkalmazása próbatétel elé állítja mind a megbízói, mind a vizsgálói oldalt.
Hosszú, kihívásokkal teli, de annál izgalmasabb tanulási folyamat áll mögöttünk, és nagy valószínűséggel hasonló vár ránk még a jövőben is, amíg az új feltételek teljeskörű tisztázása és az ezekhez való hiánytalan alkalmazkodás meg nem valósul.
Felhasznált források:
[1] Clinical Trials Regulation _ European Medicines Agency https://www.ema.europa.eu/en/human-regulatory/research-development/clinical-trials/clinical-trials-regulation
[2] Introduction to the Clinical Trials Regulation _ Deloitte Netherlands https://www2.deloitte.com/nl/nl/pages/life-sciences-en-gezondheidszorg/articles/introduction-to-the-clinical-trial-regulation.html
[3] AZ EURÓPAI PARLAMENT ÉS A TANÁCS 536/2014/EU RENDELETE https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A32014R0536
[4] AZ EURÓPAI PARLAMENT ÉS ATANÁCS 2001/20/EK IRÁNYELVE https://eur-lex.europa.eu/legal-content/EN/ALL/?uri=CELEX%3A02001L0020-20090807
A cikket összeállította:
Kenyeres Ildikó
A cikk megjelenését az MSD Pharma Hungary Kft. tette lehetővé.
HU-NON-01031
Lezárás dátuma: 2023. szeptember 7.